
Thalassemia is an inherited blood disorder that affects body’s ability to produce hemoglobin and healthy red blood cells. It affects body’s especially bone marrow ability to produce normal hemoglobin.
Thank you for reading this post, don't forget to subscribe!Pathophysiology
Hemoglobin consists of an iron-containing heme ring and four globin chains: two alpha and two non alpha. The composition of the four globin chains determines the hemoglobin type. Fetal hemoglobin (HbF) has two alpha and two gamma chains (alpha2 gamma2).
Adult hemoglobin A (HbA) has two alpha and two beta chains (alpha2 beta2), whereas hemoglobin A2 (HbA2) has two alpha and two delta chains (alpha2 delta2).
At birth, HbF accounts for approximately 80 percent of hemoglobin and HbA accounts for 20 percent. The transition from gamma globin synthesis (HbF) to beta globin synthesis (HbA) begins before birth. By approximately six months of age, healthy infants will have transitioned to mostly HbA, a small amount of HbA2, and negligible HbF.
Types of thalassemia
There are two main types of thalassemia: Alpha and beta thalassemia: Alpha and Beta Thalassemia.
Alpha Thalassemia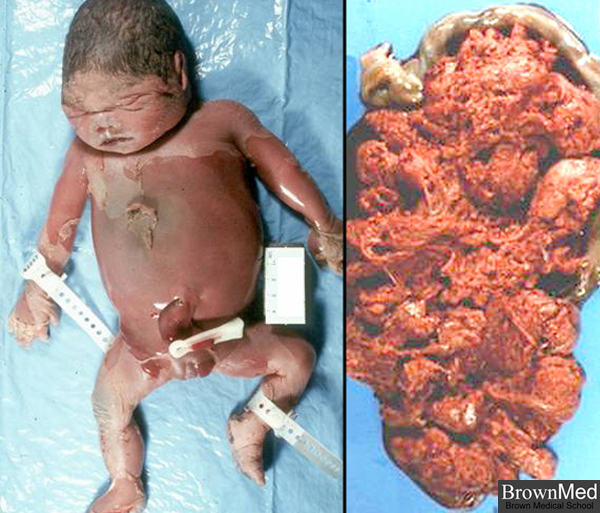
Alpha thalassemia is the result of deficient or absent synthesis of alpha globin chains, leading to excess beta globin chains.
Alpha globin chain production is controlled by two genes on each chromosome 16. Deficient production is usually caused by a deletion of one or more of these genes.
A single gene deletion results in alpha thalassemia silent carrier status, which is asymptomatic with normal hematologic findings.
The two-gene deletion causes alpha thalassemia trait (minor) with microcytosis and usually no anemia.
The three-gene deletion results in significant production of hemoglobin H (HbH), which has four beta chains (beta4).
Alpha thalassemia intermedia, or HbH disease, causes microcytic anemia, hemolysis, and splenomegaly.
The four-gene deletion results in significant production of hemoglobin Bart’s (Hb Bart’s), which has four gamma chains (gamma4).
Alpha thalassemia major with Hb Bart’s usually results in fatal hydrops fetalis.
The inheritance of alpha thalassemia is complex. Each person inherits two alpha-globin alleles from each parent. If both parents are missing at least one alpha-globin allele, their children are at risk of having Hb Bart syndrome, HbH disease, or alpha thalassemia trait.
The precise risk depends on how many alleles are missing and which combination of the HBA1 and HBA2 genes is affected.
There are four types of alpha thalassemia (as shown in table – “Prototypical forms”), hemoglobin Bart hydrops fetalis syndrome or Hb Bart syndrome (the more severe form), HbH disease, silent carrier state and trait.
Prototypical Forms of Alpha Thalassemia
| Variant | Chromosome 16 | Signs and symptoms |
|---|---|---|
| Alpha thalassemia silent carrier | One of four gene deletions | Asymptomatic |
| Alpha thalassemia trait | Two of four gene deletions | Asymptomatic |
| Hemoglobin Constant Spring | Reduced output of alpha globin | Silent or mildly symptomatic |
| Alpha thalassemia intermedia with significant hemoglobin H (hemoglobin H disease) | Three of four gene deletions | Moderate to severe hemolytic anemia, modest degree of ineffective erythropoiesis, splenomegaly, variable bone changes |
| Alpha thalassemia major with significant hemoglobin Bart’s | Four of four gene deletions | Causes nonimmune hydrops fetalis, usually fatal |
Two types of alpha thalassemia can cause health problems. The more severe type is known as hemoglobin Bart hydrops fetalis syndrome or Hb Bart syndrome. The milder form is called HbH disease.
Hb Bart syndrome is characterized by hydrops fetalis, a condition in which excess fluid builds up in the body before birth. Additional signs and symptoms can include severe anemia, an enlarged liver and spleen (hepatosplenomegaly), heart defects, and abnormalities of the urinary system or genitalia. As a result of these serious health problems, most babies with this condition are stillborn or die soon after birth. Hb Bart syndrome can also cause serious complications for women during pregnancy, including dangerously high blood pressure with swelling (preeclampsia), premature delivery, and abnormal bleeding.
HbH disease causes mild to moderate anemia, hepatosplenomegaly, and yellowing of the eyes and skin (jaundice). Some affected individuals also have bone changes such as overgrowth of the upper jaw and an unusually prominent forehead. The features of HbH disease usually appear in early childhood, but affected individuals typically live into adulthood.
Alpha thalassemia occurs frequently in people from – Mediterranean countries (Labnan, Marako, Tunzenia, Algeria, Libya, Seria, Israel, France, Italy, Cyprus, Malta, Monaco Gibraltar).
North Africa (Algeria, Morocco, Tunzenia, Egypt, libya, Western sahara, Sudan, South Sudan, Mauritania, Portugal).
Middle East (Saudi Arabia, UAE, Turkiya, Seria, Iran, Yaman, Lebnan, Quait, Bahrain, Qatar, Uman, Yaman, Iraq, Palestine, Cyprus).
South Asia (Pakistan, China, Bhutan, Bangladesh, Nepal, Afghanistan, Maldive, India, Sri Lanka).
Central Asia (Afghanistan, Kirghizstan, Turkmenistan, Kazakhstan, Uzbekistan, Tajikistan, Magnolia).
Alpha thalassemia typically results from deletions involving the HBA1 and HBA2 genes.
People who have alpha thalassemia trait can have mild anemia. However, many people with this type of thalassemia have no signs or symptoms.
Symptoms of alpha thalassemia
In patients with the characteristic features of alpha thalassemia, a reduction in the amount of normal hemoglobin prevents enough oxygen from reaching the body’s tissues. Affected individuals also have a shortage of red blood cells (anemia), which can cause pale skin, weakness, fatigue, and more serious complications.
Beta Thalassemia
Beta thalassemia is the result of deficient or absent synthesis of beta globin chains, leading to excess alpha chains. Beta globin synthesis is controlled by one gene on each chromosome 11.
Beta thalassemia occurs from any of more than 200 point mutations and (rarely) deletions of the two genes. Beta globin chain production can range from near normal to completely absent, leading to varying degrees of excess alpha globin to beta globin chain production.
The one gene defect, beta thalassemia trait (minor), is asymptomatic and results in microcytosis and mild anemia. If the synthesis from both genes is severely reduced or absent, the patient has beta thalassemia major, also known as Cooley anemia.
Patients with beta thalassemia major are almost never symptomatic at birth because of the presence of HbF, but symptoms begin to develop by six months of age. If the synthesis of beta chains is less severely reduced, the patient has beta thalassemia intermedia. These patients experience symptoms that are less severe and do not require lifelong transfusions to survive past 20 years of age.
Prototypical Forms of Beta Thalassemia
| Variant | Chromosome 11 | Signs and Symptoms |
|---|---|---|
| Beta thalassemia trait | One gene defect | Asymptomatic |
| Beta thalassemia intermedia | Two genes defective (mild to moderate decrease in beta globin synthesis) | Variable degrees of severity of symptoms of thalassemia major |
| Beta thalassemia major | Two genes defective (severe decrease in beta globin synthesis) | Abdominal swelling, growth retardation, irritability, jaundice, pallor, skeletal abnormalities, splenomegaly; requires lifelong blood transfusions6 |
Diagnosis
Most patients with thalassemia trait are found incidentally when their complete blood count shows a mild microcytic anemia. Microcytic anemia can be caused by iron deficiency, thalassemia, lead poisoning, sideroblastic anemia, or anemia of chronic disease.
The mean corpuscular volume (MCV), red blood cell distribution width (RDW), and the patient’s history can exclude some of these etiologies.
The MCV is usually less than 75 fl with thalassemia and rarely less than 80 fl in iron deficiency until the hematocrit is less than 30 percent.
For children, the Mentzer index (MCV/red blood cell count) can help distinguish between iron deficiency and thalassemia.
In iron deficiency, the ratio is usually greater than 13, whereas thalassemia yields values less than 13. A ratio of 13 would be considered uncertain.
The RDW may assist in differentiating iron deficiency and sideroblastic anemia from thalassemia. The RDW will be elevated in more than 90 percent of patients with iron deficiency, but in only 50 percent of patients with thalassemia. The RDW is usually elevated in sideroblastic anemia. Therefore, although a microcytic anemia with a normal RDW will almost always be because of thalassemia, patients with an elevated RDW will require additional testing.
Hematologic Indices of Iron Deficiency and Alpha and Beta Thalassemia
| Test | Iron deficiency | Beta thalassemia | Alpha thalassemia |
|---|---|---|---|
| MCV (abnormal if < 80 fl in adults; < 70 fl in children six months to six years of age; and < 76 fl in children seven to 12 years of age) | Low | Low | Low |
| Red blood cell distribution width | High | Normal; occasionally high | Normal |
| Ferritin | Low | Normal | Normal |
| Mentzer index for children (MCV/red blood cell count) | > 13 | < 13 | < 13 |
| Hb electrophoresis | Normal (may have reduced HbA2) | Increased HbA2, reduced HbA, and probably increased HbF | Adults: normal Newborns: may have HbH or Hb Bart’s |
Use of RDW Values in the Diagnosis of Thalassemia
Supplemental tests include serum ferritin, the peripheral smear, hemoglobin electrophoresis, serum lead level, and rarely bone marrow aspirate. Serum ferritin is the best test to screen for iron deficiency anemia. In the absence of inflammation, a normal ferritin level generally excludes iron deficiency. Serum iron, total iron-binding capacity, and transferrin saturation are rarely needed. Sideroblastic anemia can be excluded with an examination of the peripheral smear or the bone marrow aspirate. A normal serum lead level excludes lead poisoning. Anemia of chronic disease is most often a normocytic normochromic mild anemia. If thalassemia is still suspected, a hemoglobin electrophoresis may help diagnose the condition.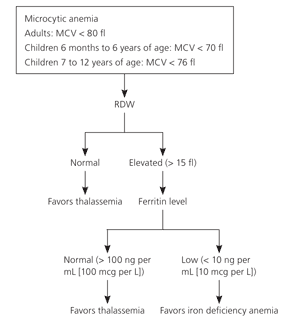
The hemoglobin electrophoresis with beta thalassemia trait usually has reduced or absent HbA, elevated levels of HbA2, and increased HbF. However, a normal concentration of HbA2 does not rule out beta thalassemia trait, especially if there was coexistent iron deficiency, which can lower HbA2 levels into the normal range.
In the newborn period, if the electrophoresis shows Hb Bart’s or HbH, the infant has alpha thalassemia. The hemoglobin electrophoresis is usually normal in adults with alpha thalassemia trait.
Patients with beta thalassemia major are diagnosed during infancy. Pallor, irritability, growth retardation, abdominal swelling, and jaundice appear during the second six months of life.
Patients with a microcytic anemia but milder symptoms that start later in life have beta thalassemia intermedia.
Also read Sickle Cell Disease or Sickle Cell Anemia (click on)
Complications
The complications that occur with beta thalassemia major or intermedia are related to overstimulation of the bone marrow, ineffective erythropoiesis, and iron overload from regular blood transfusions. Untreated infants have poor growth, skeletal abnormalities, and jaundice.
Alpha thalassemia intermedia, or HbH disease, causes hemolysis and severe anemia. Alpha thalassemia major with Hb Bart’s causes nonimmune hydrops fetalis in utero, which is almost always fatal.
With multiple blood transfusions and continued absorption of intestinal iron, iron overload develops. Iron is deposited in visceral organs (mainly the heart, liver, and endocrine glands), and most patient deaths are caused by cardiac complications. Endocrinopathies, particularly hypogonadism and diabetes mellitus, may occur in adolescents and adults.
Splenomegaly invariably develops in the symptomatic thalassemias. Splenomegaly can worsen the anemia and occasionally cause neutropenia and thrombocytopenia.
Thromboembolic events, venous and arterial, are not uncommon. Persons with beta thalassemia major or intermedia may develop a chronic hypercoagulable state, especially after splenectomy.
Osteopenia and osteoporosis are being found more often as persons with thalassemia major live longer. One study found osteoporosis in 51 percent of persons older than 12 years with thalassemia major.
Allopathic Treatment for Thalassemia
Blood Transfusions
Patients with beta thalassemia major require periodic and lifelong blood transfusions to maintain a hemoglobin level higher than 9.5 g per dL (95 g per L) and sustain normal growth. The need for blood transfusions may begin as early as six months of age. For patients with beta thalassemia intermedia, the decision to transfuse is a more subjective clinical assessment.
Transfusion requirements are episodic and become necessary when the patient’s hemoglobin is inadequate for a normal life or when the anemia impairs growth and development.
Alpha thalassemia intermedia, or HbH disease, causes mild to moderate hemolysis. Transfusions will occasionally be necessary depending upon the severity of the clinical condition.
Chelation (Bonding)
Transfusion-dependent patients develop iron overload because they have no physiologic process to remove excess iron from multiple transfusions. Therefore, they require treatment with an iron chelator starting between five and eight years of age.
Deferoxamine, subcutaneously or intravenously, has been the treatment of choice. Although this therapy is relatively less toxic, it is cumbersome and expensive. Adverse effects of deferasirox were transient and gastrointestinal.
Bone marrow transplant
Bone marrow transplantation in childhood is the only allopathic curative therapy for beta thalassemia major. Hematopoietic stem cell transplantation generally results in an excellent outcome in low-risk patients, defined as those with no hepatomegaly, no portal fibrosis on liver biopsy, and regular chelation therapy, or at most, two of these abnormalities.
thalassemia major. Hematopoietic stem cell transplantation generally results in an excellent outcome in low-risk patients, defined as those with no hepatomegaly, no portal fibrosis on liver biopsy, and regular chelation therapy, or at most, two of these abnormalities.
Management of Specific Conditions
If hypersplenism causes a marked increase in transfusion requirements, splenectomy may be needed. Surgery is usually delayed until at least four years of age because of the spleen’s role in clearing bacteria and preventing sepsis. At least one month before surgery, patients should receive the pneumococcal polysaccharide vaccine. Children should also receive the pneumococcal conjugate vaccine series. Antibiotic prophylaxis with penicillin, 250 mg orally twice a day, is recommended for all persons during the first two years after surgery and for children younger than 16 years.
Endocrinopathies
Growth hormone therapy for beta thalassemia intermedia and major has had variable success and is not generally recommended. Hormone therapy is effective for hypogonadism.
Pregnancy
Preconception genetic counseling is strongly advised for all persons with thalassemia. Two parents, each with beta thalassemia trait, have a one in four chance of conceiving a child with beta thalassemia major and a three in four chance the child will have thalassemia trait or be normal (Online Figure A).
Patients with alpha thalassemia trait have a more complex pattern of inheritance. Whether both defective genes are on the same or different chromosomes will alter the outcome (Online Figure B).
Chorionic villus sampling using polymerase chain reaction technology to detect point mutations or deletions can identify infants affected with beta thalassemia.
Patients with alpha thalassemia trait should consider prenatal diagnosis because Hb Bart’s increases the risk of toxemia and postpartum bleeding. Preimplantation genetic diagnosis is becoming available in conjunction with in vitro fertilization.
Cardiac
Serum ferritin has been used as a marker of iron storage to predict cardiac complications. Ferritin levels less than 2,500 ng per mL (2,500 mcg per L) are associated with improved survival. However, ferritin levels are unreliable when liver disease is present.
Hypercoagulopathy
No randomized or observational studies regarding anticoagulation, antiplatelet therapy, or both have been reported for persons at greater risk of thromboembolic events. Therefore, no specific treatment can be recommended. Persons with a history of thrombosis may be treated with low-molecular-weight heparin. Anticoagulation therapy is warranted before surgery and during pregnancy for at-risk persons. Alternatives to estrogen-containing contraception should be offered to women of reproductive age.
both have been reported for persons at greater risk of thromboembolic events. Therefore, no specific treatment can be recommended. Persons with a history of thrombosis may be treated with low-molecular-weight heparin. Anticoagulation therapy is warranted before surgery and during pregnancy for at-risk persons. Alternatives to estrogen-containing contraception should be offered to women of reproductive age.
Psychosocial
Beta thalassemia major or intermedia is a chronic disease with a significant impact on the patient and the patient’s family and offspring. Education about the genetics of the disease, prenatal diagnosis options, and psychological therapy to help manage the complications is appropriate. However, neither the type of education nor the duration of therapy can be specified based on current evidence.
Vitamin Deficiencies
Folic acid deficiency has been reported in thalassemia major and intermedia as a result of increased erythropoiesis. Therefore, daily oral supplementation with 1 mg of folic acid is recommended for persons with evidence of folate deficiency.
Because some complications seem to be related to cellular oxidative stress, treatment with antioxidants has been thought to be beneficial. However, no improvements in anemia or reductions in morbidity or mortality have been demonstrated. Vitamin C is not recommended except in transfusion-dependent patients with a proven deficiency.
Prognosis With allopathic treatment
Patients with thalassemia trait have a normal life expectancy. Persons with beta thalassemia major live an average of 17 years and usually die by 30 years of age. Most deaths are caused by the cardiac complications of iron overload.
Homeopathic treatment for thalassemia
For the treatment of thalassemia, homeopathic medicines are prescribed only on the basis of TOTALITY OF SYMPTOMS with a clear cut identification of disease.
Here are few homeopathic medicines for thalassemia with good and promising results:
Chininum Sulphuricum
Angina pectoris. Asthma. Brow ague. Cancers. Haematuria. Haemoglobinuria. Haemorrhages. Headache. Intermittent fever. Spleen, enlarged. Typhus-fever. Face pale colour, sickly look, sunken eyes. Earth-coloured face. The white of the eyes discoloured and eyes dull. Complexion icteric. Cyanosis. Erosion of the gums, and of the wall of the buccal cavity, with violent pain and gangrenous crusts. Accumulation of mucus in the mouth, with nocturnal angina, augmented secretion of saliva. Bitter taste of mouth. Nausea with risings (empty or bitter). Hepatomegaly. Splenomegaly.. White stools. Aching pains. Darting and incisive pains. Pulsating, tensive, burning, and expansive pains. Gangrenous inflammations. Sensation of coldness with internal tremblings. Limbs weakness – trembling.
Haemorrhages. Headache. Intermittent fever. Spleen, enlarged. Typhus-fever. Face pale colour, sickly look, sunken eyes. Earth-coloured face. The white of the eyes discoloured and eyes dull. Complexion icteric. Cyanosis. Erosion of the gums, and of the wall of the buccal cavity, with violent pain and gangrenous crusts. Accumulation of mucus in the mouth, with nocturnal angina, augmented secretion of saliva. Bitter taste of mouth. Nausea with risings (empty or bitter). Hepatomegaly. Splenomegaly.. White stools. Aching pains. Darting and incisive pains. Pulsating, tensive, burning, and expansive pains. Gangrenous inflammations. Sensation of coldness with internal tremblings. Limbs weakness – trembling.
Natrum Muriaticum
A great remedy for certain forms of intermittent fever, anaemia, chlorosis, many disturbances of the alimentary tract and skin. Great debility; most weakness felt in the morning in bed. Coldness. Emaciation most notable in neck. Great liability to take cold. Dry mucous membranes. Constrictive sensation throughout the body. Great weakness and weariness. Oversensitive to all sorts of influences. Hyperthyroidism. Goitre. Addison’s disease. Diabetes. Hungry, yet lose weight. Tachycardia. Sensation of coldness of heart. Heart and chest feel constricted. Fluttering, palpitating; intermittent pulse. Heart’s pulsations shake body. Palms hot and perspiring. Arms and legs (knees) feel weak. Hangnails. Dryness and cracking about fingernails. Numbness and tingling in fingers and lower extremities. Dry eruptions. Crusty eruptions. Hydraemia in chronic malarial states with weakness, constipation, loss of appetite, etc. Sweats on every exertion. Sickle Cell Disease. Sickle Cell Anemia
Ferrum Metallicum
Weakness, pale, anaemic and chlorotic, with pseudo-plethora. Pallor of skin, mucous membranes, face, alternating with flushes. Stinging headache. Face white, bloodless and puffy. Voracious appetite, or absolute loss of appetite. Vomiting immediately after eating. Undigested stools. Tachicardia. Anaemic murmur.
Phosphorus
Paralysis. Destroyed weak bone, especially the lower jaw and tibia. Fatty degeneration of blood vessels and every tissue and organ of the body. Haemorrhages, and haematogenous jaundice. Yellow atrophy of the liver (cirrhosis) and sub-acute hepatitis. Liver congested. Acute hepatitis. Fatty degeneration. Jaundice. Pancreatic disease. Large, yellow spots on abdomen. Sudden prostration, faints, sweats, shooting pains. Polycythemia (high number of red blood cells in the blood). Muscular pseudohypertrophy, neuritis. Inflammation of the respiratory tract. Paralytic symptoms. Tertiary syphilis, skin lesions, and nervous debility. Scurvy. Pseudo-hypertrophic paralysis. Ataxia and adynamia. Osteomyelitis. Bone fragility. Violent heart palpitation with anxiety. Pulse rapid, small, and soft. Heart dilated. Feeling of warmth in heart.
Calcarea Arsenicosa
Chronic malaria. Infantile enlarged liver and spleen. Nephritis, with great sensitiveness in kidney region. slightest emotion causing palpitation. Dyspnoea, with feeble heart. Chilliness. Albuminuria. Dropsy. Affections of spleen and mesenteric glands. Hemoglobin and red corpuscles are low. Confusion, delusions, illusions. Great depression. Enlarged liver and spleen in children. Pancreatic disease; relieves burning pain in pancreatic cancer. Belching with saliva and beating of heart. Constriction and cardiac pain, suffocating feeling, palpitation, oppression and throbbing. Cancers. Inflammation in veins of lower extremities. Weariness and lameness of lower limbs.
Lachesis Mutus
Trifacial neuralgia. Tearing pain in jaw-bones. Face pale purple, mottled, puffed; looks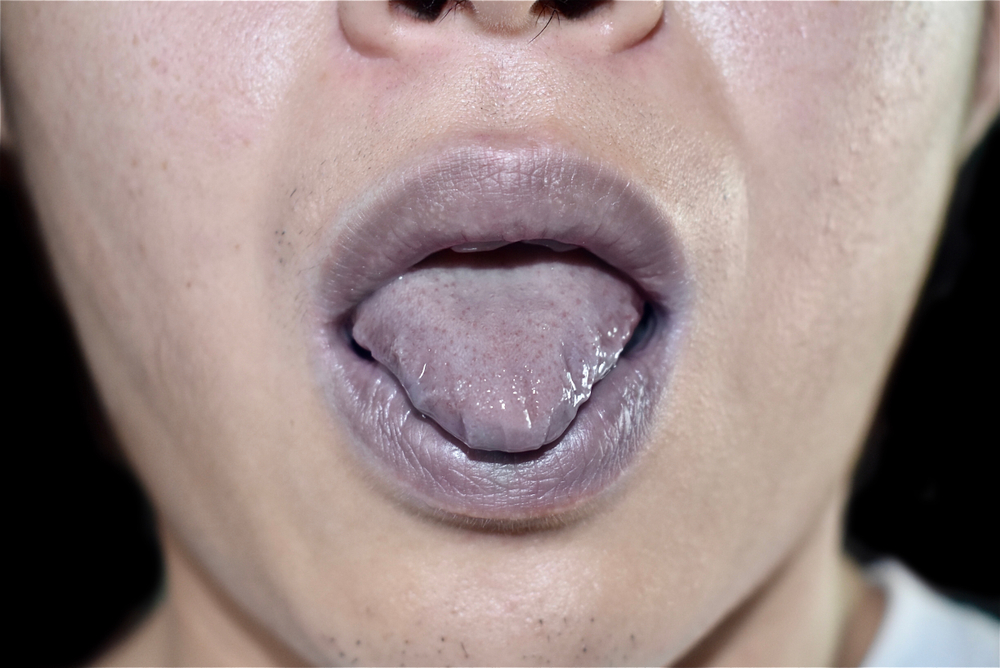 swollen, bloated, jaundiced, cyanotic. Gums swollen, spongy, bleed. Tongue swollen, burns, trembles, red, dry and cracked at tip. Abdomen tympanitic, sensitive, painful. Hepatomegaly. Splenomegaly. Constricted feeling causing bradycardia with anxiety. Cyanosis. Irregular beats. Hot perspiration, bluish, purplish appearance. Boils, carbuncles, ulcers. Blue-black swellings. Pyemia; dissecting wounds. Purpura, with intense prostration. Senile erysipelas. Wens. Cellulitis. Varicose ulcers.
swollen, bloated, jaundiced, cyanotic. Gums swollen, spongy, bleed. Tongue swollen, burns, trembles, red, dry and cracked at tip. Abdomen tympanitic, sensitive, painful. Hepatomegaly. Splenomegaly. Constricted feeling causing bradycardia with anxiety. Cyanosis. Irregular beats. Hot perspiration, bluish, purplish appearance. Boils, carbuncles, ulcers. Blue-black swellings. Pyemia; dissecting wounds. Purpura, with intense prostration. Senile erysipelas. Wens. Cellulitis. Varicose ulcers.
Plumbum metallicum
The great choice of medicine for general sclerotic conditions; The blood, alimentary and nervous systems are the special seats of action of Plumbum. Lead paralysis of extensors, forearm or upper limb, from center to periphery with partial anaesthesia or excessive hyperesthesia, preceded by pain. Localized neuralgic pains, neuritis. Hematosis is interfered with, rapid reduction in number of red corpuscles; hence pallor, icterus, anaemia. Constrictive sensation in internal organs. Delirium, coma and convulsions. Hypertension and arteriosclerosis. Progressive muscular atrophy. Infantile paralysis. Locomotor ataxia. Excessive and rapid emaciation. Bulbar paralysis. Important in peripheral affections. The points of attack for Plumbum are the neurexins and the anterior horns. Multiple sclerosis, posterior spinal sclerosis. Contractions and boring pain. Nephritis with amaurosis and cerebral symptoms. Pale and cachectic. Yellow, corpse-like; cheeks sunken. Skin of face greasy, shiny. Tremor of naso-labial muscles. Gums swollen, pale; distinct blue lines along margins of gums. Tongue tremulous, red on margin. Constant vomiting. Cardiac weakness. Pulse soft and small, dichroite. Wiry pulse, camp-like constriction of peripheral arteries. ellow, dark-brown liver spots. Jaundice. Dry. Dilated veins of forearms and legs.
Thiosinaminum
Scar tissue, tumors, enlarged glands; lupus, strictures, adhesions. Ectropion, opacities of cornea, cataract, ankylosis, fibroids, scleroderma. Tinnitus. Lightning pains. Gastric, vesicle and rectal crises. Arterio-sclerotic vertigo.
Antipyrinum
Induced leukocytosis. Dilated capillaries of skin. Patches of hyperaemia and swelling. Sickle Cell Disease or Sickle Cell Anemia
Arsenicum Album
A profoundly acting medicine on every organ and tissue. Prevailing debility, exhaustion, and restlessness, with nightly aggravation. Great exhaustion after the slightest exertion. Irritable weakness. Burning pains. Unquenchable thirst. Burning relieved by heat. Fear fright and worry. Green discharges. Infantile visceral leishmaniasis . Ptomaine poisoning, stings, dissecting wounds. Anaemia and chlorosis. Degenerative changes. Gradual loss of weight. Reduces the refractive index of blood serum. Maintains the system under the stress of malignancy regardless of location. Malarial cachexia. Septic infections and low vitality. Sickle Cell Disease. Sickle Cell Anemia.
Arsenicum Iodatum
Influenza, hay-fever, old nasal catarrhs, and catarrh of middle ear. Swelling of tissues within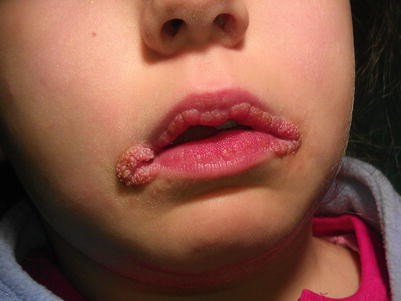 the nose. Hypertrophic condition of eustachian tube and deafness. Senile heart, myocarditis and fatty degeneration. Pulse shotty. Chronic aortitis. Epithelioma of the lip. Cancers. Pain and pyrosis. Vomiting an hour after food. Nausea distressing. Pain in epigastrium. Slight hacking cough, with dry and stopped-up nostrils. Pleuritis exudativa. Chronic bronchitis. Pulmonary tuberculosis. Pneumonia that fails to clear up. Drenching night-sweats. Pulse rapid, feeble, weak, irregular. Ichthyosis. Enlarged scrofulous glands. Venereal bubo. Debilitating night-sweats. Profound prostration, rapid, irritable pulse, recurring fever and sweats, emaciation; tendency to diarrhoea. Chronic pneumonia, with abscess in lung. Hectic; debility; night sweats. Cardiac weakness, emaciation and general debility; in chronic, watery diarrhoea in phthisical patients. Anaemic palpitation and dyspnoea. Chronic pneumonia. Great emaciation. Arteriosclerosis, myocardial degeneration and senile heart. Threatened pyaemia.
the nose. Hypertrophic condition of eustachian tube and deafness. Senile heart, myocarditis and fatty degeneration. Pulse shotty. Chronic aortitis. Epithelioma of the lip. Cancers. Pain and pyrosis. Vomiting an hour after food. Nausea distressing. Pain in epigastrium. Slight hacking cough, with dry and stopped-up nostrils. Pleuritis exudativa. Chronic bronchitis. Pulmonary tuberculosis. Pneumonia that fails to clear up. Drenching night-sweats. Pulse rapid, feeble, weak, irregular. Ichthyosis. Enlarged scrofulous glands. Venereal bubo. Debilitating night-sweats. Profound prostration, rapid, irritable pulse, recurring fever and sweats, emaciation; tendency to diarrhoea. Chronic pneumonia, with abscess in lung. Hectic; debility; night sweats. Cardiac weakness, emaciation and general debility; in chronic, watery diarrhoea in phthisical patients. Anaemic palpitation and dyspnoea. Chronic pneumonia. Great emaciation. Arteriosclerosis, myocardial degeneration and senile heart. Threatened pyaemia.
China Officianalis
Debility from exhausting discharges, from loss of vital fluids, together with a nervous erethism. Blue color around eyes. Hollow eyes. Yellowish sclerotica. Black specks, bright dazzling illusions. Vomiting of undigested food. Slow digestion. Darting pain crosswise in hypogastric region. Tympanitic abdomen. Pain in right hypochondrium. Gall-stone colic. Liver and spleen swollen and enlarged. Jaundice. Internal coldness of stomach and abdomen. Undigested, frothy, yellow, painless stool. Heart irregular with weak rapid beats followed by strong, hard beats. Suffocative attacks, syncope. Pains in limbs and joints.
Natrum sulphuricum
Asthma. Biliousness. Condylomata. Debility. Diabetes. Dyspepsia. Enuresis. Epilepsy. Epistaxis (menstrual). Fistulous abscesses. Gonorrhoea. Headache. Hydraemia. Influenza. Leukemia. Hepatomegaly. Malaria. Migraine. Nephritic scarlatina. Ophthalmia. Panaritium. Phlegmasia alba dolens. Photophobia. Phthisis. Scrofulous ophthalmia. Spleen affections. Sycosis. Face pale and sickly. Breath shortness. Painful sensibility of limbs, which feel as if bruised, fatigued. Prostration, tired. Limbs trembling – weakness. Itching, and itching pimples, eczemas.
Kali sulphuricum
Suppressed rash of measles. Abundant scaling of epidermis. Burning itching, papular eruption exuding pus-like moisture. Fine red pimples running together. Scurf, scaling, chapping. Sores with yellow, sticky secretions. Epithelial cancer. Eczema. Itch. Intertrigo. Nails diseased. Jaundice. Hard, tympanitic abdomen – Hepatomegaly.
Ceanothus Americanus
This remedy seems to possess a specific relation to the spleen. Ague cake of malaria. Anaemic patients where liver and spleen are at fault. Chronic bronchitis with profuse secretion. Marked blood pressure, reducing powers. Active hemostatic, materially reducing blood clotting. Enormous enlargement of the spleen. Spleen pain. Deep-seated pain in left hypochondrium, hypertrophy of spleen. Leukemia. Violent dyspnoea. Menses profuse, and yellow weakening leucorrhoea. Unable to lie on left side. Pain in liver and back.
Tinospora Cordifolia or Guduchi Giloy (Guduchi), Giloy (Pakistan).
Fevers. Debility. Dyspepsia. Jaundice. Skin diseases. Diarrhoea and dysentery. Leprosy; helminthiasis and rheumatoid arthritis. Splenic affections. Secondary syphilis. Debility caused by repeated attacks of fever. Intermittent fever. Acute and chronic malarial fevers. Bilious vomiting associated with thirst and headache. Enlargement of liver and spleen. Excessive tachycardia.
Vanadium Metallicum
Its action is that of an oxygen carrier and a catalyzer. Increases amount of hemoglobin, also combines its oxygen with toxines and destroys their virulence. Also increases and stimulates phagocytes. A remedy in degenerative conditions of the liver and arteries. Anorexia and symptoms of gastro intestinal irritation; albumen, casts and blood in urine. Tremors; vertigo; hysteria and melancholia; neuroretinitis and blindness. Anaemia, emaciation. Cough dry, irritating and paroxysmal, sometimes with haemorrhages. Irritation of nose, eyes and throat. Tuberculosis, chronic rheumatism, diabetes. Acts as a tonic to digestive function and in early tuberculosis. Arteriosclerosis, sensation as if heart was compressed, as if blood had no room in the aorta. Anxious pressure on whole chest. Fatty heart. Degenerative states, has brain softening. Atheroma of arteries of brain and liver.
For consultation; Feel free to whatsapp us or visit our clinic.
P. S : This article is only for doctors having good knowledge about Homeopathy and allopathy, for learning purpose(s).
For proper consultation and treatment, please visit our clinic.
Location, address and contact numbers are given below.
NoN of above mentioned medicine(s) is/are the full/complete treatment, but just hints for treatment; every patient has his/her own constitutional medicine.
To order medicine by courier, please send your details at WhatsApp– +923119884588
 Dr. Sayyad Qaisar Ahmed (MD {Ukraine}, DHMS), Abdominal Surgeries, Oncological surgeries, Gastroenterologist, Specialist Homeopathic Medicines.
Dr. Sayyad Qaisar Ahmed (MD {Ukraine}, DHMS), Abdominal Surgeries, Oncological surgeries, Gastroenterologist, Specialist Homeopathic Medicines.
Senior research officer at Dnepropetrovsk state medical academy Ukraine.
Location: Al-Haytham clinic, Umer Farooq Chowk Risalpur Sadder (0923631023, 03119884588), K.P.K, Pakistan.
Find more about Dr Sayed Qaisar Ahmed at :
https://www.youtube.com/Dr Qaisar Ahmed